This page is outdated, please also see the man page.
How to run fdensity?
This mode allows measuring the density of functional annotations around the positions of a collection of QTLs. To illustrate how it works, first download the required example data:
- The results from a full cis-analysis: TXT
- The functional annotation for few Transcription Factor Binding sites: BED
Step1: Prepare the QTL data
First, you need to prepare a BED file containing the positions of the QTLs of interest. To do so, extract all significant hits at a given FDR threshold (e.g. 5%):
Rscript ./script/runFDR_cis.R results.genes.full.txt.gz 0.05 results.genes
Then, transform the significant QTL list into a BED file using the following command:
cat results.genes.significant.txt | awk '{ print $9, $10-1, $11, $8, $1, $5 }' | tr " " "\t" | sort -k1,1 -k2,2n > results.genes.significant.bed
The resulting BED file looks like this:
head results.genes.significant.bed
1 15210 15211 1_15211 ENSG00000227232.4 -
1 735984 735985 1_735985 ENSG00000177757.1 +
1 735984 735985 1_735985 ENSG00000240453.1 -
1 739527 739528 1_739528 ENSG00000237491.4 +
1 754963 754964 1_754964 ENSG00000225880.4 -
1 769222 769223 1_769223 ENSG00000228794.4 +
1 832397 832398 1_832398 ENSG00000230699.2 +
1 844342 844343 1_844343 ENSG00000223764.2 -
1 879675 879676 1_879676 ENSG00000187634.6 +
1 895705 895706 1_895706 ENSG00000187961.9 +
Note here that this file contains the follwing columns:
- 1. Chromosome ID
- 2. Start position of the QTL
- 3. End position of the QTL
- 4. QTL ID
- 5. Targeted phenotype ID (not used, can be whatever you want)
- 6. Strand orientation; this is important, you need to specify here on which strand the QTL has been mapped to
Step2: Run the density measurements
Now that you've got your QTLs ready to go, you can then measure the density using the following command:
QTLtools fdensity --qtl results.genes.significant.bed --bed TFs.encode.bed.gz --out density.TF.around.QTL.txt
This generates a file density.TF.around.QTL.txt that contains the number of functional annotations per 1kb bins around QTLs.
head density.TF.around.QTL.txt
-1000000 -999001 1314
-999000 -998001 1253
-998000 -997001 1263
-997000 -996001 1315
-996000 -995001 1339
-995000 -994001 1293
-994000 -993001 1277
-993000 -992001 1299
-992000 -991001 1309
-991000 -990001 1282
- 1. start position of the bin
- 2. end position of the bin
- 3. number of annotations in this bin
You can use the two following options to tune the calculations:
- --bin [int]: size of the bin in bp (default is 1000bp)
- --window [int]: size of the window around QTLs to be examined in bp (default is 1000000bp)
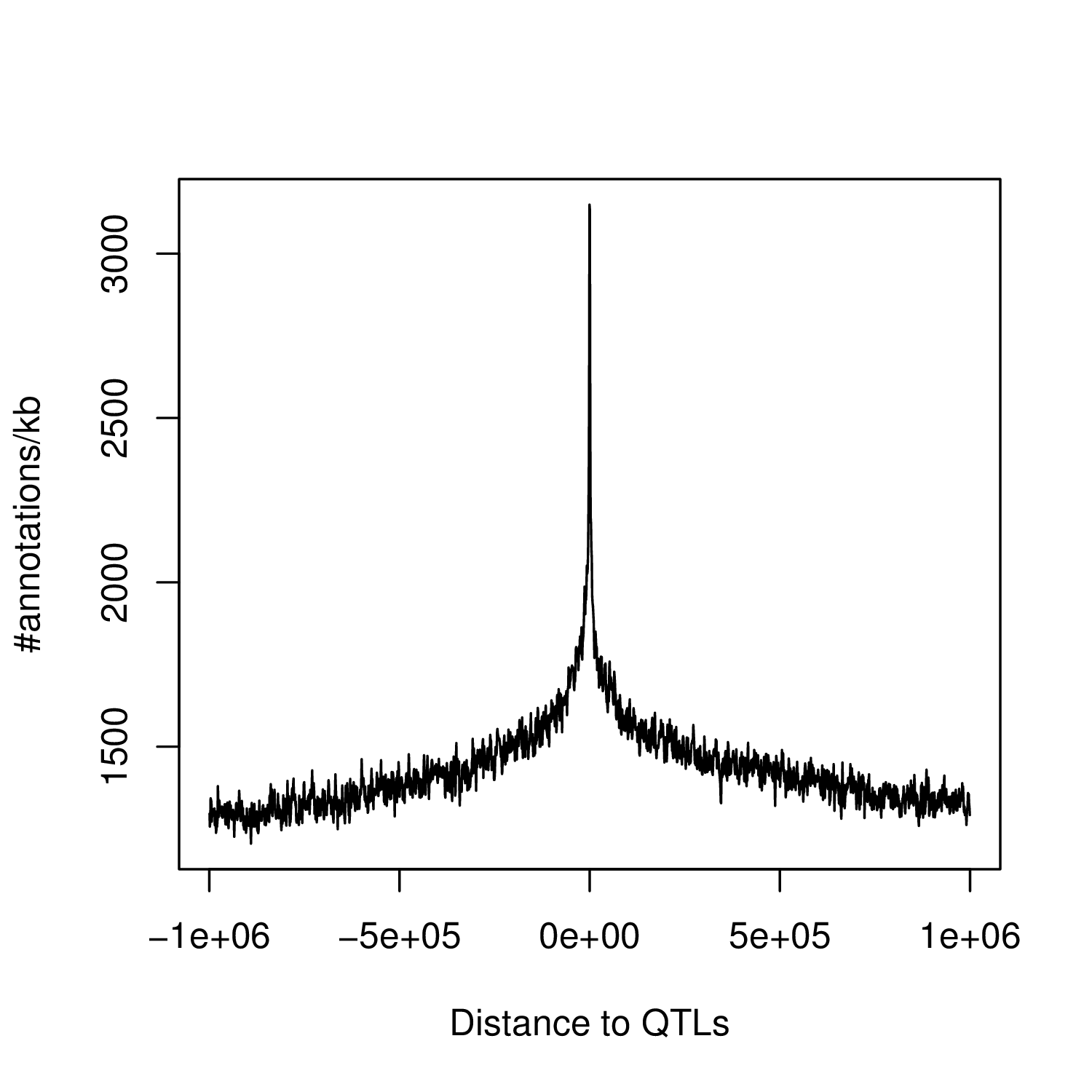
Once you've got your density measurements, you can plot them in R using:
R> D=read.table("density.TF.around.QTL.txt", head=FALSE, stringsAsFactors=FALSE)
R> plot((D$V1+D$V2)/2, D$V3, type="l", xlab="Distance to QTLs", ylab="#annotations/kb")
This is going to give you something like this: